制备型(Prep)液相色谱(LC)是采用色谱方法,将目标化合物从原料、副反应或其它杂质中分离出一定量达到足够纯度的化合物,以用于后续实验或处理的色谱方法。
使用OBD制备级方法转换计算器弥补分析级到制备级应用之间的性能差距
https://www.waters.com/waters/promotionDetail.htm?id=134713448&locale=101
包括:
Waters梯度计算器-由分析柱条件转换到制备柱条件https://www.waters.com/webassets/other/lp/prep_calc/AnalyticaltoPrep/NewPrepcalculator.htm
比较柱压和载样负荷https://www.waters.com/webassets/other/lp/prep_calc/ColumnBackpressures/PrepMass.htm
UPLC转换聚焦梯度换算https://www.waters.com/webassets/other/lp/prep_calc/UPLCtoPrepFocusCalc/UPLCtoPrepFocusCalc.htm
转换用计算软件https://www.waters.com/waters/support.htm?lid=134891632&type=DWNL
柱选择工具https://find.waters.com/ColumnCoach/allselectivity/getchart
液相色谱纯化法:LC纯化系统的配置与一般液相色谱系统相同,但增加了馏分收集器。样品混合物经进样装置定量由流动相传输至色谱柱,色谱柱根据组分特有的化学或物理性质对其进行分离。由检测器检出组分,系统根据设置将流动样品输送至废液,或者收集器。收集操作中,检测器信号会触发馏分收集器将液流输送至收集容器中。输送至收集容器的馏分纯度取决于分离过程中化合物与其它邻近洗脱杂质的分离度。
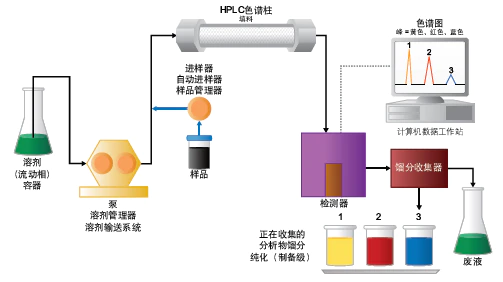
图1:制备型LC系统
判断纯化分离成功与否的标准包括通量、回收率和所得分离物的纯度。
纯化分离由色谱峰的分离或“分离度”驱动。
以下是对分离度影响最为显著的色谱参数:
§ 色谱柱填料(固定相)
§ 溶剂强度
§ 溶剂类型
§ 缓冲液添加剂
§ 柱温
执行方法开发工作基本流程如下图所示:
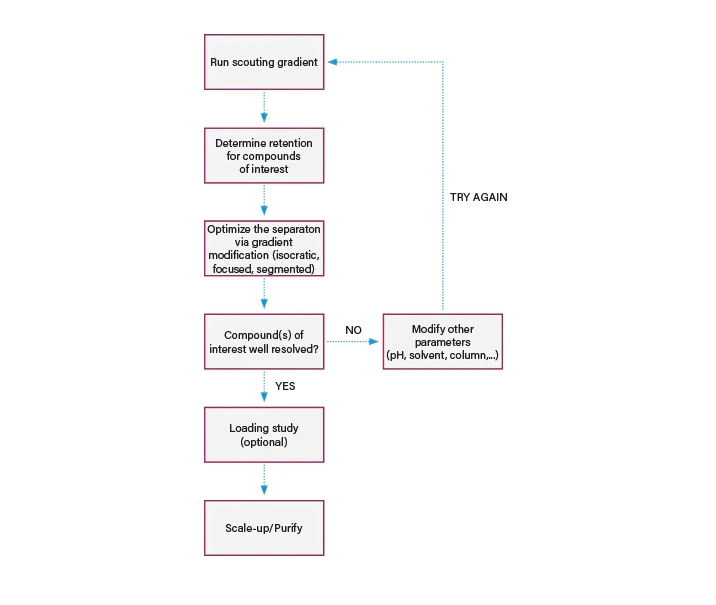
图2:通用制备型工作流程
在设计工作流程之前,十分重要的考虑因素是目标化合物的性质。
如果从熟知的来源或新来源中分离已知化合物,很容易通过检索相关文献获取有关目标化合物色谱行为的信息,并从已发表的方法中选择适用的分离方法。
对于含有未知类型化合物的粗提取物,设计分离方案的难度较大。在这种情况下,可在初始分离之后进行一系列探索性实验,获取更多有关目标化合物的信息,例如pKa、分子量、溶解性、稳定性、UV光谱和生物活性等。然后根据这些信息修改初始分离方法,使其符合化合物的化学和物理性质要求。这些信息不仅有助于方法开发,还可以在收集到馏分之后,为掌握目标产物的稳定性提供非常大的帮助。
第一步要制备样品并使用快速探索梯度执行常规分离。这一步通常为小规模分离,目的是节省宝贵的样品。根据该分离的结果,可以计算洗脱目标化合物的溶剂条件并进一步优化分离条件,尽可能提高分离度。此外,可能还需要对上样量进行研究,确定可维持适当分离度以实现高纯度分离的理想上样量。
确定分离方法和理想上样量后,通常要针对具有应用前景或潜在价值的分离物进行方法放大(但并不是必须执行)。术语“规模”(scale)描述满足产物纯度、通量和收率等应用目标所需的仪器和方法。若要“放大”(scaled-up)方法,则需要将方法转换至硬件配置可处理更高上样量的系统。从本质上来讲,方法放大就是借助一系列方法放大计算和硬件更改,在分离度保持不变的前提下,将方法从分析级内径色谱柱转换到制备级内径色谱柱的过程。

图3:用于分析级和制备级纯化的色谱柱内径对比
样品
可用于分离特定化合物的样品来源十分广泛,包括药物中间体、天然产物、保健食品、饮料和工业产品等。只要原料可以进行提取并溶解于液体基质中,就可以使用色谱纯化其中的各组分。
样品提取技术的选择取决于待提取原料的复杂性。对于天然产物,通常将样品干燥并研磨成细颗粒用以提高提取效率。接下来采用渗滤(用于大体积样品)或浸提(用于小体积样品)等技术提取目标化合物。这两种技术都需要向样品原料中加入溶剂,然后在超声、涡旋或浸泡后收集溶质。收集提取物之后,和所有HPLC样品一样,样品在进样前必须过滤,以除去颗粒并排除气泡。
分离模式
制备型色谱主要有四种分离模式:反相、正相、凝胶渗透和离子交换。合适的分离模式取决于待分析样品、提取物或混合物与固定相和溶剂之间的相容性。
反相色谱是开发纯化方法时最常用的技术,其使用极性弱于洗脱溶剂极性的固定相。反相色谱使用水与乙腈或甲醇的混合物作为洗脱液,其中还会添加缓冲液(酸或碱)用于控制样品的离子化程度以及结合固定相中未反应的游离硅醇基团,可减少峰拖尾并提高色谱重现性。
快速启动方法开发
建立反相分离方法时,通常选择小规格的分析型色谱柱(内径≤ 4.6 mm),并且其柱长和填料应可扩展至较大规格(内径≥ 10 mm)内径的色谱柱,以便将来放大方法。方法开发的关键是找到可实现出色分离的适用溶剂体系。对于酸性化合物,通常采用pH为酸性的水性流动相,并以甲醇或乙腈作为强溶剂。浓度为0.1%的甲酸是常用的缓冲液添加剂,因为其兼容UV和质谱检测。如需鉴定未知化合物或获取纯分离物用于进一步研究,MS兼容性将非常有用。如需让碱性分子保持未电离状态,可使用pH为碱性而非酸性的流动相。使用任何pH的缓冲液之前,都应参阅色谱柱固定相说明书了解其兼容性。水性流动相通常由位置A处的泵泵送,强溶剂由位置B处的泵泵送。本指南将分别使用“A”和“B”指代泵表中的水性溶剂和强溶剂。

1 目的 规范TENSOR 27红外光谱仪的操作程序,正确使用仪器,保证设备安全和检测的顺利进行。 2 使用环境 电源电压:85~265V,47~65Hz 温度范...

同一种微生物在不同的国家或地区常有不同的名称,这就是俗名。俗名在局部地区可以使用,但不便于交流,容易引起混乱。为在世界范围...

人是能够思想的苇草 帕斯卡尔 人只不过是一根苇草,是自然界最脆弱的东西;但他是一根能思想的苇草。用不着整个宇宙都拿起武器来才能...

以下内容转载自小木虫 哥特复兴VS [1]Han-Dong Sun,Yi-Ming Shi:LCUV-Guided Isolation and Structure Determination of Lancolide E: A Nortriterpenoid with a Tetracyclo [...


