BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。BLAST程序能迅速与公开数据库进行相似性序列比较。BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:
1、BLASTP是蛋白序列到蛋白库中的一种查询。库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
NCBI的在线blast:http://blast.ncbi.nlm.nih.gov/Blast.cgi
1,进入在线blast界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。不同的blast程序上面已经有了介绍。这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。选择一个要比对的数据库。关于数据库的说明请看NCBI在线blast数据库的简要说明。一般的话参数默认。
3,blast参数的设置。注意显示的最大的结果数跟E值,E值是比较重要的。筛选的标准。最后会说明一下。
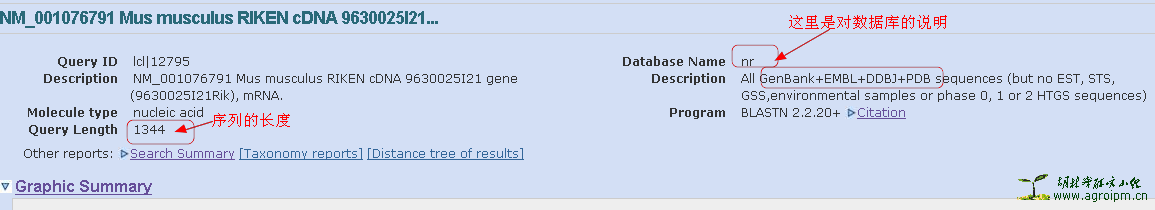
4,注意一下你输入的序列长度。注意一下比对的数据库的说明。
5,blast结果的图形显示。没啥好说的。
6,blast结果的描述区域。注意分值与E值。分值越大越靠前了,E值越小也是这样。

7,blast结果的详细比对结果。注意比对到的序列长度。评价一个blast结果的标准主要有三项,E值(Expect),一致性(Identities),缺失或插入(Gaps)。加上长度的话,就有四个标准了。如图中显示,比对到的序列长度为1405,看Identities这一值,才匹配到1344bp,而输入的序列长度也是为1344bp(看上面的图),就说明比对到的序列要长一点。由Qurey(起始1)和Sbjct(起始35)的起始位置可知,5'端是是多了一段的。有时也要注意3'端的。
附:
E值(Expect):表示随机匹配的可能性,E值越大,随机匹配的可能性也越大。E值接近零或为零时,具本上就是完全匹配了。
一致性(Identities):或相似性。匹配上的碱基数占总序列长的百分数。
缺失或插入(Gaps):插入或缺失。用"—"来表示。

GB /T 7 714《文后参考文献著录规则》是一项专门供著者和编辑编撰文后参考文献使用的国家标准,非等效采用ISO 690《文献工作文后参考文献...
一、参考文献是对期刊论文引文进行统计和分析的重要信息源之一 ,在本规范中采用 GB 7714推荐的顺序编码制编排。 二、参考文献著录项目...
参考文献是科技论文的重要组成部分,也是编辑加工和重要内容。温哥华格式要求,著录文后参考文献时,英文刊名和人名一律用缩写。这...
The following come fromhttp://www.smartdraw.com SmartDraw Software, LLC Since 1994, SmartDraws purpose has been to expand the ways in which people communicate so that we can clearly...







